Software
HaMMLET: Dynamically compressed Bayesian Hidden Markov Model using Haar Wavelet Shrinkage
HaMMLET is a powerful open-source implementation of a Bayesian Hidden Markov Model. It uses the Haar wavelet transform to dynamically compress the data, which leads to improved speed and convergence of Forward-Backward Gibbs Sampling. It can be used in applications such as CNV detection from aCGH data.
Visit HaMMLET's main website at http://wiedenhoeft.github.io/HaMMLET/. More...
Gato: Graph Animation Toolbox

Gato - the Graph Animation Toolbox - is a software which visualizes algorithms on graphs. Graphs are mathematical objects consisting of vertices and edges connecting pairs of vertices: think of cities as vertices and interstates as edges connecting two cities. Algorithms might find a shortest path - the fastest route -- or a minimal spanning tree or solve one of other interesting problems on graphs: maximal-flow, weighted and non-weighted matching and min-cost flow. Visualization means linking cause - the statements of an algorithm - immediately to an effect - changes to the graph the algorithm has as its input - by terms of blinking, changing colors and other visual effects.
Visit Gato's main website at http://gato.sf.net/. More...
dvstar: dvstar: Construction and comparison of variable-length Markov chains
dvstar is an open-source tool for constructing and applying variable-length Markov chains (VLMCs) as applied to genomes. The VLMCs are computed based on k-mer counts, enabling them to be built on arbitrarily large genomes and sequencing data. Using the computed VLMCs, the similarity between VLMCs can be assessed using either the dvstar comparison method, which compares parameters of the models or with the negative log-likelihood to assess similarity to other genomes. The tool has been used to study the evolution of viruses and can be used to monitor environments for emerging pathogens. More...
Past Software Projects
MVQueries: Classifying short gene expression time-courses
Short gene expression time-courses monitoring response to toxins are represented as piecewise constant functions, which are modeled as left–right Hidden Markov Models. Our software implements a Bayesian approach to parameter estimation and in inference. Compared to previously published work, we improve prediction accuracy by 7 and 4%, respectively, when classifying toxicology and stress response data and e also reduce running times by at least a factor of 140. More...
ClusterViz: Cluster Visualisation

ClusterViz is a software to visualize the clustering process using the family of k-means algorithms
Visit ClusterViz's main website at http://clusterviz.sourceforge.net/. More...
TreqCG: Clustering Accelerates High-Throughput Sequencing Read Mapping
As high-throughput sequencers become standard equipment outside of sequencing centers, there is an increasing need for efficient methods for pre-processing and primary analysis. While a vast literature proposes methods for NGS data analysis, we argue that significant improvements can still be gained by exploiting expensive pre-processing steps which can be amortized with savings from later stages. More...
Tileomatic: Design of oligonucleotide arrays
Tileomatic is a software to design optimal spaced oligonucleotide tiling arrays. Tileomatic balances the three main conflicting objectives in tiling array design—oligonucleotide probe spacing, probe quality and hybridization conditions—to arrive at a globally optimal solution. It is most effective for spaced tiling arrays where variations in spacing can reduce variations in hybridization conditions and avoid having to use low-quality of cross-hybridizing probes. Candidate oligonucleotide probe sets are pre-computed with our OSProbes software
Visit Tileomatic's main website at http://tileomatic.molgen.mpg.de/. More...
GHMM: General Hidden Markov Model library
<p>The General Hidden Markov Model library (GHMM) is a freely available LGPL-ed C library implementing efficient data structures and algorithms for basic and extended HMMs. The development is hosted at Sourceforge <a HREF="http://sourceforge.net/projects/ghmm/">http://sourceforge.net/projects/ghmm/</a>, where you have access to the Subversion repository, mailing lists and forums.</p>
Visit GHMM's main website at http://ghmm.org/. More...
GQL: Graphical Query Language

GQL is a suite of tools for analyizing time-course experiments. Currently, it is adapted to gene expression data. The two main tools are GQLQuery, for querying data sets, and GQLCluster, which provides a way for computing groupings based on a number of methods (model-based clustering using HMMs as cluster models and estimation of a mixture of HMMs).
Visit GQL's main website at http://ghmm.org/gql. More...
Turtle: Identifying frequent k-mers with cache-efficient algorithms
Counting the frequencies of k-mers in read libraries is often a first step in the analysis of high-throughput sequencing experiments. Counting frequencies of large read libraries like human can be very time and memory intensive. We present a novel method that balances time, space and accuracy requirements to efficiently extract frequent k-mers. More...
TreQ: Indel-tolerant read mapping
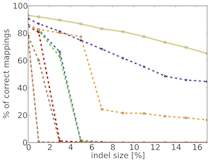
TreQ is a read mapper for high-throughput DNA sequencing reads, in particular reads from 100 nt to hundreds of nucleotides, and for large edit distance between sequencing read and match in the reference genome. In contrast to existing read mappers, TreQ can cope particularly well with indels, either one long indel; see the figure giving the percentage of accurate matches as a function of indel length for 200 nt reads. TreQ performs best at a time comparable to BWA at large edit distance settings, SSAHA2 is the second best but is five times slower than tree. This makes TreQ an excellent choice for analyzing genetic variants in low-coverage situations and without the need for paired-end sequencing. TreQ will be released under the GPL upon publication.
Visit TreQ's main website at http://treq.sf.net/. More...
MCPD: Markov Chain Pooling Decoder
The Markov Chain Pooling Decoder (MCPD) is used in the analyis of pooling experiments for library screening. Pools are collections of clones, and screening a pool with a probe is a group test, determining whether any of these clones are positive for the probe. The results of the pool screenings are interpreted, or decoded, to infer which clones are candidates to be positive using a Markov chain Monte Carlo approach. MCPD implements this MCMC to compute marginal probabilities of clones using a Bayesian model for the experiment. More...
Proclust: Protein clustering by transitive homology
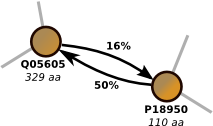
Proclust is software package for clustering protein sequences with a graph-based approach which significantly increases the numbers of remote homolog proteins detected. You can use the online server at the ZAIK, University of Cologne or download the software. Proclust is released under the GPL. More...
SLiQ: Simple linear inequalities based Mate-Pair reads filtering and scaffolding

Mate pair filtering is an import pre-processing step for contig scaffolding. SLiQ inequalities have been shown to be a much better filter than traditional majority voting based filter. Our software applies the inequalities and filters the Mate pairs. It then produces a contig graph and applies the Naive scaffolding algorithm described in the paper (http://bioinformatics.rutgers.edu/Static/Publications/SLIQ_arxiv.pdf). More...
PBQ: The Python Batch Queue
PBQ is a simple batch queue system, with the goal of completing a list of jobs on a bunch of machines with a shared file system without interfering with interactive users and/or more important batch jobs. Most importantly, you do not need to be root to install or use it. PBQ is distributed under the GNU Public License (GPL). More...
PyMix: The Python mixture package
The Python Mixture Package is a freely available Python library implementing algorithms and data structures for a wide variety of data mining applications with basic and extended mixture models.
Visit PyMix's main website at http://www.pymix.org. More...
pGQL: probabilistic Graphical Query Language

pGQL is a software tool in particular for analyzing gene expression time courses. It allows its user to interactively define linear HMM queries on time course data using rectangular graphical widgets called probabilistic time boxes. The analysis is fully interactive and the graphical display shows the time courses along with the graphical query. The results can be submitted to gPROF directly from pGQL. More...